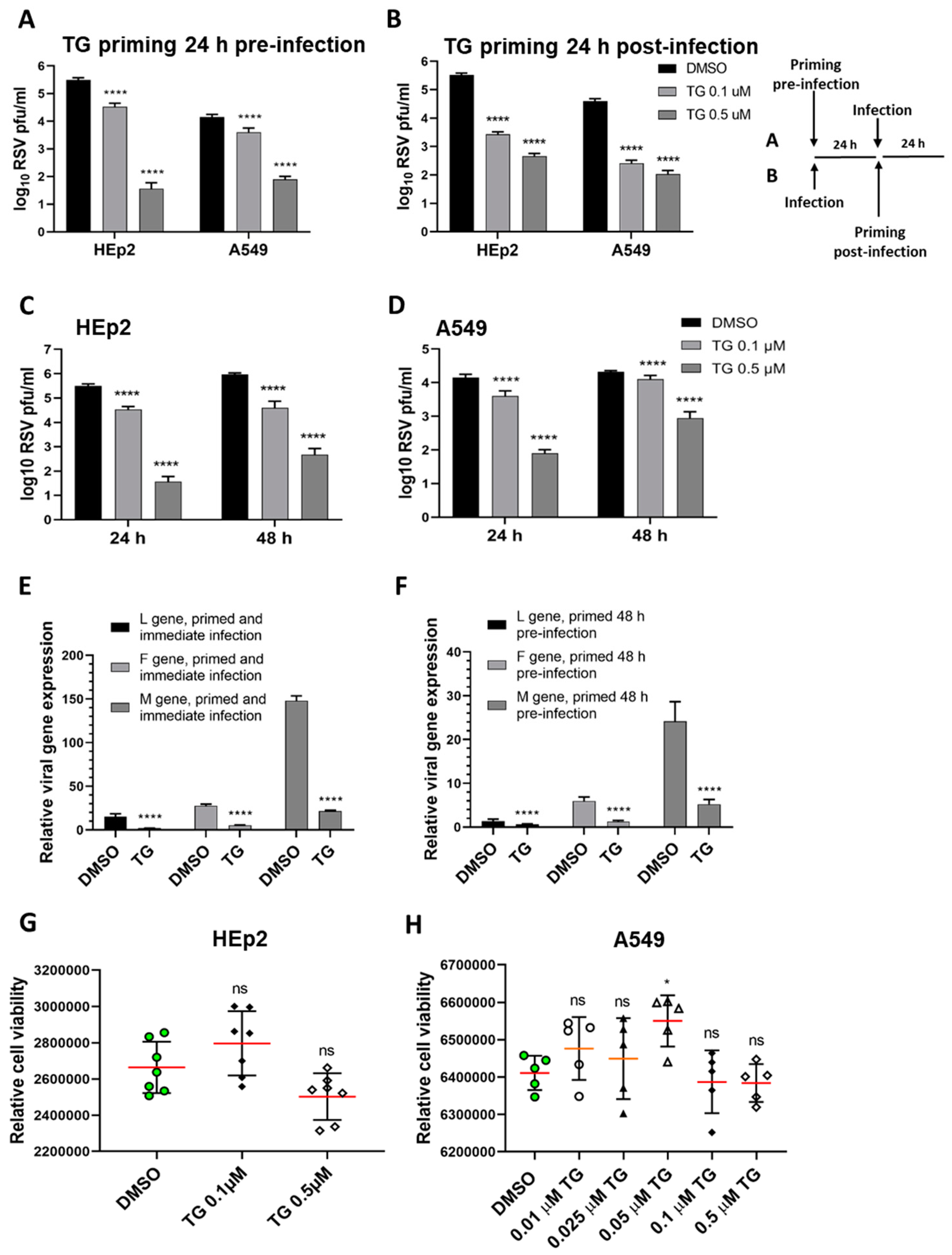
Figure 1.
Short exposure (30 min) of thapsigargin (TG) to human cells, at non-cytotoxic levels, promptly elicits a prolonged (≥48 h) antiviral state that blocks RSV replication. TG priming 24 h before (A) or 24 h after (B) infection, in a dose-dependent manner, blocks RSV production. (A) HEp2 and A549 were primed with TG or control DMSO for 30 min, washed with PBS and incubated in normal culture media for 24 h before RSV infection, or (B) were initially infected with RSV for 24 h before 30 min of TG priming. The TG-induced antiviral state lasted at least 48 h. HEp2 (C) and A549 (D) cells were primed with TG or DMSO control for 30 min, as indicated, washed with PBS and allowed a further period of 24 or 48 h of normal culture; after which cells were infected with RSV. All cells were infected with RSV (A2 strain, ATCC VR-1540) at 0.1 MOI for a total duration of 3 days. The spun supernatants were collected to infect HEp2 cells for 24 h for immuno-detection of RSV with mouse anti-RSV (2F7) antibody (pfu/mL). Significance by 2-way ANOVA (Sidak’s multiple comparisons) is relative to the corresponding DMSO control. HEp2 cells were more permissive to RSV replication than A549 cells. TG inhibited RSV transcription. HEp2 cells were (E) primed for 30 min with 0.5 µM TG immediately before RSV infection, and (F) primed with TG for 30 min, washed with PBS and cultured for a further period of 48 h before RSV infection. After a total of 3 days of infection, total RNA was extracted for cDNA conversion to quantify viral gene (L-gene, M-gene and F-gene) expression normalised to 18s rRNA. Significance was determined by unpaired t-test relative to expression in the corresponding DMSO control. (G) HEp2 and (H) A549 cells pre-incubated with indicated concentrations of TG or DMSO control for 30 min were washed with PBS, cultured overnight in serum-free media (Opti-MEM) and subjected to cell viability assays (CellTiter-Glo® Luminescent Cell Viability Assay kit, Promega). Horizontal bars = mean (red) ± standard deviation; ns = not significant. Significance by one-way ANOVA with Dunnett’s multiple comparisons is relative to the corresponding DMSO control. All assays were in triplicates and were performed three times. * p < 0.05 and **** p < 0.0001.
Figure 1.
Short exposure (30 min) of thapsigargin (TG) to human cells, at non-cytotoxic levels, promptly elicits a prolonged (≥48 h) antiviral state that blocks RSV replication. TG priming 24 h before (A) or 24 h after (B) infection, in a dose-dependent manner, blocks RSV production. (A) HEp2 and A549 were primed with TG or control DMSO for 30 min, washed with PBS and incubated in normal culture media for 24 h before RSV infection, or (B) were initially infected with RSV for 24 h before 30 min of TG priming. The TG-induced antiviral state lasted at least 48 h. HEp2 (C) and A549 (D) cells were primed with TG or DMSO control for 30 min, as indicated, washed with PBS and allowed a further period of 24 or 48 h of normal culture; after which cells were infected with RSV. All cells were infected with RSV (A2 strain, ATCC VR-1540) at 0.1 MOI for a total duration of 3 days. The spun supernatants were collected to infect HEp2 cells for 24 h for immuno-detection of RSV with mouse anti-RSV (2F7) antibody (pfu/mL). Significance by 2-way ANOVA (Sidak’s multiple comparisons) is relative to the corresponding DMSO control. HEp2 cells were more permissive to RSV replication than A549 cells. TG inhibited RSV transcription. HEp2 cells were (E) primed for 30 min with 0.5 µM TG immediately before RSV infection, and (F) primed with TG for 30 min, washed with PBS and cultured for a further period of 48 h before RSV infection. After a total of 3 days of infection, total RNA was extracted for cDNA conversion to quantify viral gene (L-gene, M-gene and F-gene) expression normalised to 18s rRNA. Significance was determined by unpaired t-test relative to expression in the corresponding DMSO control. (G) HEp2 and (H) A549 cells pre-incubated with indicated concentrations of TG or DMSO control for 30 min were washed with PBS, cultured overnight in serum-free media (Opti-MEM) and subjected to cell viability assays (CellTiter-Glo® Luminescent Cell Viability Assay kit, Promega). Horizontal bars = mean (red) ± standard deviation; ns = not significant. Significance by one-way ANOVA with Dunnett’s multiple comparisons is relative to the corresponding DMSO control. All assays were in triplicates and were performed three times. * p < 0.05 and **** p < 0.0001.
![Viruses 13 00234 g001]()
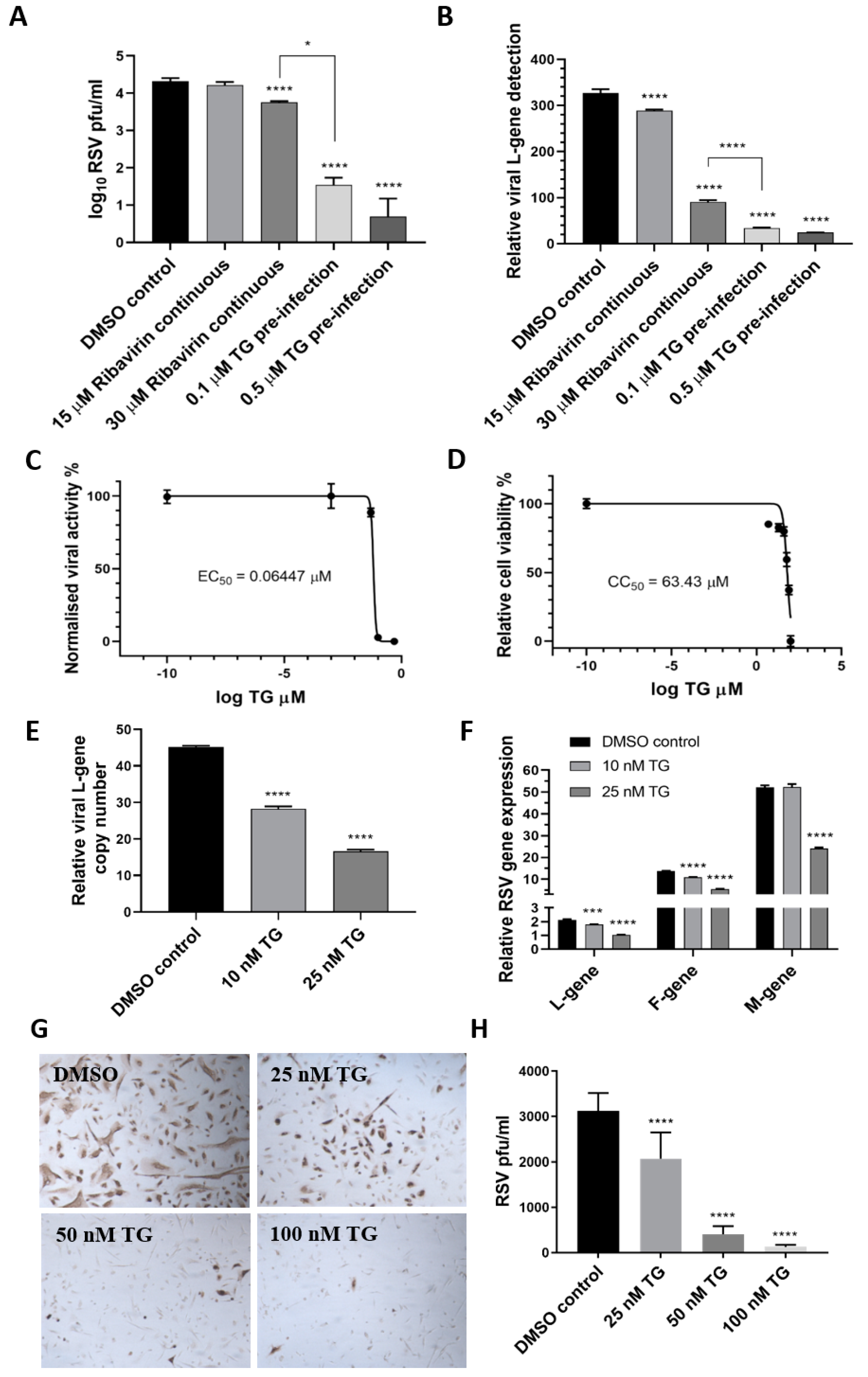
Figure 2.
TG is more effective as an antiviral than ribavirin, shows a high selectivity index and blocks viral transcription and viral protein production. HEp2 cells were primed with TG, ribavirin or DMSO control, as indicated, for 30 min, washed with PBS and infected with RSV, as earlier described, in the continuous presence of ribavirin in media or in media alone (for TG or DMSO control). Media were harvested at 4 dpi for (A) progeny virus titration (pfu/mL) and (B) detection of viral L-gene RNA by one-step reverse transcription-qPCR. Unless otherwise indicated, significance by one-way ANOVA (Tukey’s multiple comparisons) is relative to the DMSO control. The corresponding selectivity index (SI) of TG in RSV infection of HEp2 cells is estimated at 984 (C,D). The effective concentration (EC) and cytotoxic concentration (CC) of TG against RSV in HEp2 cells were determined by pfu/mL virus titrations and luminescence cell viability assays, respectively, over a pre-infection priming range of TG. SI = CC50/EC50 = 63.43/0.06447 = 983.9. EC90 = 84.55 nM. NHBE cells were primed with TG or the DMSO control, as indicated, for 30 min, washed with PBS and infected with RSV at 0.1 MOI. At 48 hpi, media were harvested for the detection of the viral L-gene RNA by one-step reverse transcription-qPCR (E), and total RNA was extracted for cDNA conversion to quantify the expression of viral genes (L-gene, F-gene and M-gene) normalised to 18s rRNA (F). Significance by one-way ANOVA (Dunnett’s multiple comparisons) (E) and by 2-way ANOVA (Tukey’s multiple comparisons) (F) is relative to the DMSO control. TG also inhibited viral F-protein production in NHBE cells. Cells were primed with TG or the DMSO control, as indicated, for 30 min, washed with PBS and infected with RSV at 0.05 MOI for 48 h and directly immunostained for the presence of RSV F-protein (G,H). Images captured at 100 times magnification. There was a clear reduction in the number of RSV-positive cells (pfu) with an increasing priming dose of TG. Significance by one-way ANOVA (Dunnett’s multiple comparisons) is relative to the DMSO control. * p < 0.05, *** p < 0.001 and **** p < 0.0001.
Figure 2.
TG is more effective as an antiviral than ribavirin, shows a high selectivity index and blocks viral transcription and viral protein production. HEp2 cells were primed with TG, ribavirin or DMSO control, as indicated, for 30 min, washed with PBS and infected with RSV, as earlier described, in the continuous presence of ribavirin in media or in media alone (for TG or DMSO control). Media were harvested at 4 dpi for (A) progeny virus titration (pfu/mL) and (B) detection of viral L-gene RNA by one-step reverse transcription-qPCR. Unless otherwise indicated, significance by one-way ANOVA (Tukey’s multiple comparisons) is relative to the DMSO control. The corresponding selectivity index (SI) of TG in RSV infection of HEp2 cells is estimated at 984 (C,D). The effective concentration (EC) and cytotoxic concentration (CC) of TG against RSV in HEp2 cells were determined by pfu/mL virus titrations and luminescence cell viability assays, respectively, over a pre-infection priming range of TG. SI = CC50/EC50 = 63.43/0.06447 = 983.9. EC90 = 84.55 nM. NHBE cells were primed with TG or the DMSO control, as indicated, for 30 min, washed with PBS and infected with RSV at 0.1 MOI. At 48 hpi, media were harvested for the detection of the viral L-gene RNA by one-step reverse transcription-qPCR (E), and total RNA was extracted for cDNA conversion to quantify the expression of viral genes (L-gene, F-gene and M-gene) normalised to 18s rRNA (F). Significance by one-way ANOVA (Dunnett’s multiple comparisons) (E) and by 2-way ANOVA (Tukey’s multiple comparisons) (F) is relative to the DMSO control. TG also inhibited viral F-protein production in NHBE cells. Cells were primed with TG or the DMSO control, as indicated, for 30 min, washed with PBS and infected with RSV at 0.05 MOI for 48 h and directly immunostained for the presence of RSV F-protein (G,H). Images captured at 100 times magnification. There was a clear reduction in the number of RSV-positive cells (pfu) with an increasing priming dose of TG. Significance by one-way ANOVA (Dunnett’s multiple comparisons) is relative to the DMSO control. * p < 0.05, *** p < 0.001 and **** p < 0.0001.
![Viruses 13 00234 g002]()
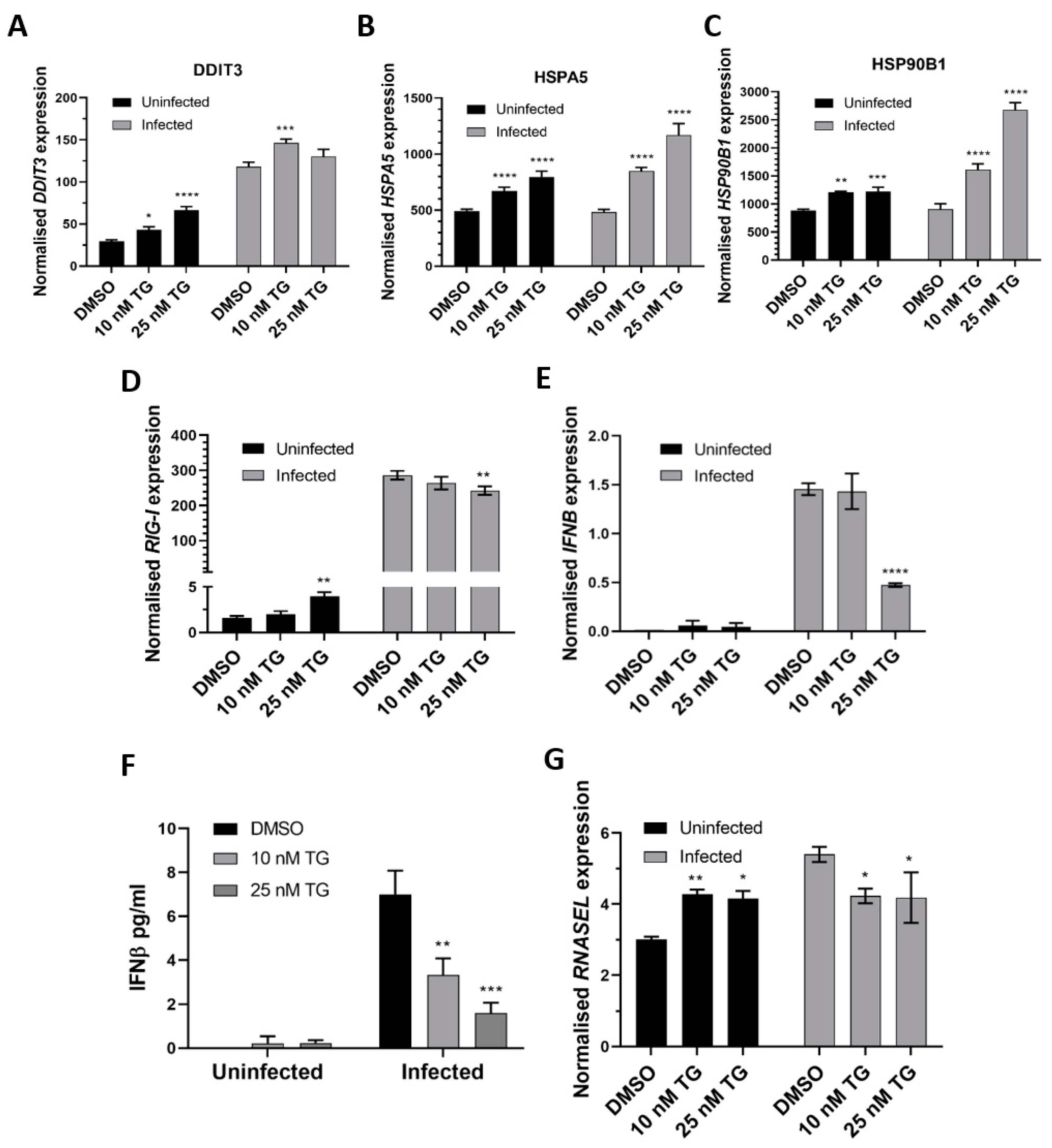
Figure 3.
TG priming of NHBE cells increases expression of the ER stress genes (pre- and post-infection) and basal expression of the RIG-I signalling-associated genes, but during infection, induction of the RIG-I-associated genes is attenuated. Cells were primed with TG or DMSO control for 30 min, washed with PBS and infected with RSV at 0.05 MOI 48 h after which total RNA was extracted for cDNA conversion to quantify expression of the ER stress genes (A) DDIT3, (B) HSPA5 and (C) HSP90B1. Expression was normalised to 18s rRNA and the indicated significance, determined by 2-way ANOVA (Sidak’s multiple comparisons), is relative to the corresponding DMSO control. There was consistent indication of pre-infection activation of the RIG-I-associated genes, RIG-I (D), IFNB (E) (and corresponding IFNB protein, (F)) and RNASEL (G) from TG priming. IFNB ELISA was performed on supernatants at 16 hpi. All RNA expression was normalised to 18s rRNA and the indicated significance, determined by one-way ANOVA and Dunnett’s multiple comparisons (D), or 2-way ANOVA Tukey’s multiple comparisons (E–G), is relative to the corresponding DMSO control. All assays were in triplicates and were performed three times. * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
Figure 3.
TG priming of NHBE cells increases expression of the ER stress genes (pre- and post-infection) and basal expression of the RIG-I signalling-associated genes, but during infection, induction of the RIG-I-associated genes is attenuated. Cells were primed with TG or DMSO control for 30 min, washed with PBS and infected with RSV at 0.05 MOI 48 h after which total RNA was extracted for cDNA conversion to quantify expression of the ER stress genes (A) DDIT3, (B) HSPA5 and (C) HSP90B1. Expression was normalised to 18s rRNA and the indicated significance, determined by 2-way ANOVA (Sidak’s multiple comparisons), is relative to the corresponding DMSO control. There was consistent indication of pre-infection activation of the RIG-I-associated genes, RIG-I (D), IFNB (E) (and corresponding IFNB protein, (F)) and RNASEL (G) from TG priming. IFNB ELISA was performed on supernatants at 16 hpi. All RNA expression was normalised to 18s rRNA and the indicated significance, determined by one-way ANOVA and Dunnett’s multiple comparisons (D), or 2-way ANOVA Tukey’s multiple comparisons (E–G), is relative to the corresponding DMSO control. All assays were in triplicates and were performed three times. * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
![Viruses 13 00234 g003]()
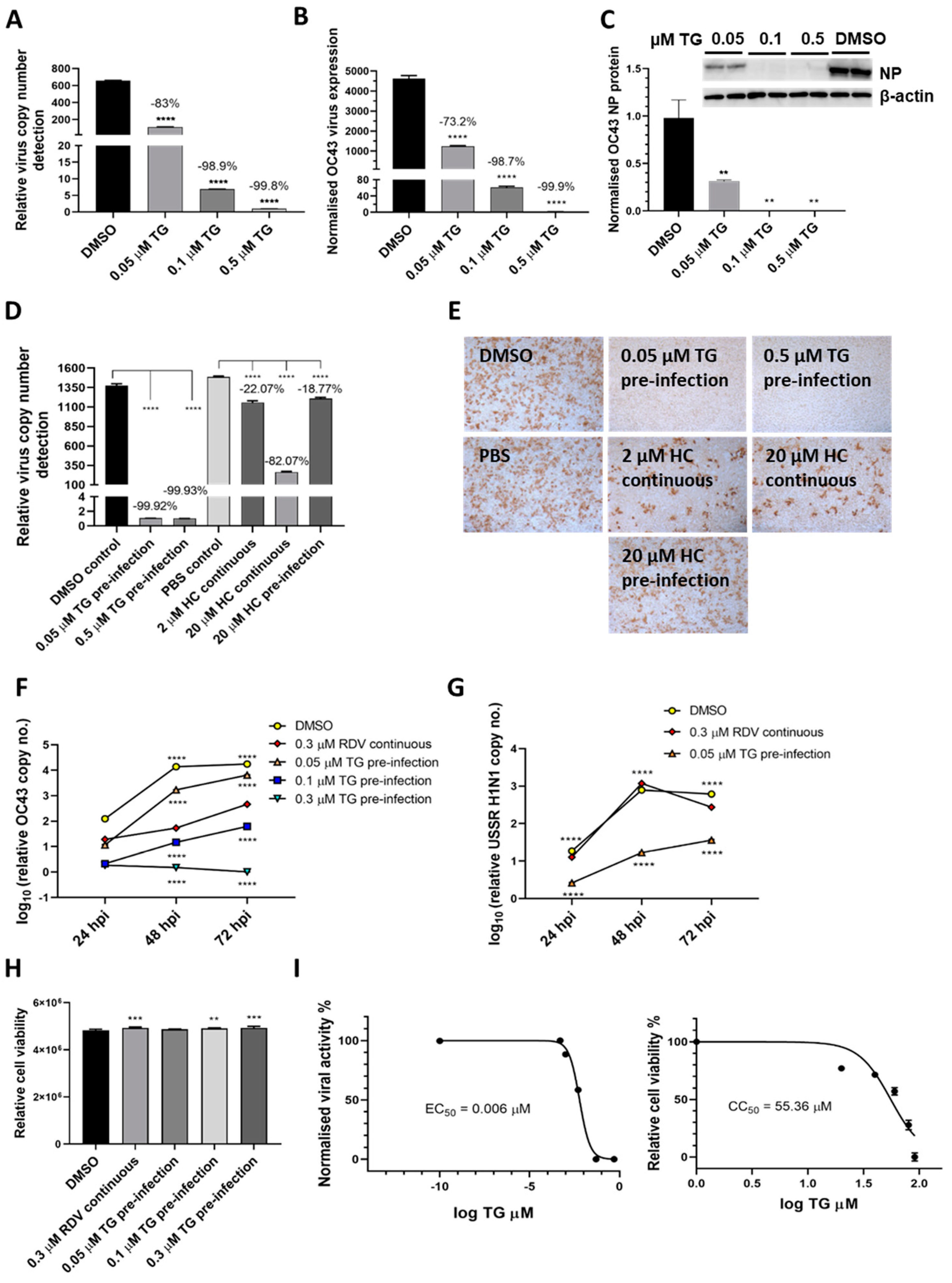
Figure 4.
TG inhibits OC43 viral transcription and protein production, is more effective as an antiviral than hydroxychloroquine and remdesivir, and exhibits a high selectivity index. (A,B) A549 cells were primed with TG for 30 min, washed twice with PBS and infected with OC43 at 0.5 MOI for 3 h; cells were then washed again with PBS and cultured for 48 h in serum-free media (Opti-MEM supplemented with 0.1 µg/mL TPCK trypsin), after which RNA was extracted from (A) culture media and (B) cells for one-step reverse transcription qPCR, and cDNA conversion followed by qPCR, respectively, to detect viral OC43 replicase polyprotein 1ab RNA; cDNA quantification was normalised to 18s rRNA. (C) Duplicate set of wells were used for cellular protein extractions to detect OC43 NP by Western blotting. (D,E) MRC5 cells were primed with TG, hydroxychloroquine (HC) or the DMSO/PBS control, as indicated, for 30 min, washed twice with PBS, infected with OC43 (at 0.01 MOI) for 3 h, further washed with PBS twice and finally replenished with serum-free media in the absence of a compound (pre-infection) or continued presence of HC (continuous). At 2 dpi, media were collected for (D) detection of viral polyprotein 1ab RNA by one-step reverse transcription-qPCR and (E) direct progeny virus detection by infecting A549 cells for 24 h and immunostaining for the presence viral NP. Images captured at 100 times magnification. The indicated significance (determined by one-way ANOVA) and percentage reduction in viral RNA detection are relative to the corresponding controls. (F,G) TG was superior to remdesivir (RDV) in blocking OC43 (F) and influenza A virus (G) replication. The A549 cells were primed with the indicated TG, 0.3 µM RDV or DMSO control for 30 min, washed twice with PBS and infected with 0.01 MOI of CoV OC43 or 1.0 MOI of USSR H1N1 virus for 2 h, after which cells were washed again with PBS and incubated in serum-free media for TG primed cells, or in media in the continuous presence of RDV. At 24, 48 and 72 hpi, viral RNA extraction was performed on the collected supernatants followed by one-step reverse transcription qPCR to detect the relative copy number of OC43 replicase polyprotein 1ab RNA or influenza M-gene RNA, based on the relative Ct method. The indicated significance is relative to the corresponding RDV-treated cells based on 2-way ANOVA Dunnett’s (F) or Tukey’s (G) multiple comparisons test. (H) RDV and TG treatments had no adverse effect on cell viability. A549 cells were treated continuously with RDV, or for 30 min with the indicated TG or DMSO, washed, cultured overnight and subjected to cell viability assay (CellTiter-Glo 2.0 Cell Viability Assay, Promega). The indicated significance was determined by one-way ANOVA relative to the DMSO control. (I) Selectivity index (CC50/EC50) of TG in OC43 inhibition was estimated at between 7072 and 9227. EC90 = 0.02622 μM. MRC5 cells were primed with TG (0 to 91 µM) for 30 min, washed twice with PBS and culture in DMEM Glutamax with 10% FCS and 1% P/S overnight. Cell viability assay (CC50) was performed with CellTiter-Glo 2.0 Cell Viability Assay (Promega). The effective or inhibition TG dose response (EC50) was based on priming of MRC5 cells with the indicated concentrations of TG (0 to 0.5 µM) for 30 min followed by PBS washing and infection with OC43 at 0.01 MOI. Three days post-infection, the supernatants were harvested for RNA extraction and one-step reverse-transcription qPCR to quantify the presence of viral RNA (polyprotein 1ab RNA). CC = cell cytotoxicity; EC = effective concentration. ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
Figure 4.
TG inhibits OC43 viral transcription and protein production, is more effective as an antiviral than hydroxychloroquine and remdesivir, and exhibits a high selectivity index. (A,B) A549 cells were primed with TG for 30 min, washed twice with PBS and infected with OC43 at 0.5 MOI for 3 h; cells were then washed again with PBS and cultured for 48 h in serum-free media (Opti-MEM supplemented with 0.1 µg/mL TPCK trypsin), after which RNA was extracted from (A) culture media and (B) cells for one-step reverse transcription qPCR, and cDNA conversion followed by qPCR, respectively, to detect viral OC43 replicase polyprotein 1ab RNA; cDNA quantification was normalised to 18s rRNA. (C) Duplicate set of wells were used for cellular protein extractions to detect OC43 NP by Western blotting. (D,E) MRC5 cells were primed with TG, hydroxychloroquine (HC) or the DMSO/PBS control, as indicated, for 30 min, washed twice with PBS, infected with OC43 (at 0.01 MOI) for 3 h, further washed with PBS twice and finally replenished with serum-free media in the absence of a compound (pre-infection) or continued presence of HC (continuous). At 2 dpi, media were collected for (D) detection of viral polyprotein 1ab RNA by one-step reverse transcription-qPCR and (E) direct progeny virus detection by infecting A549 cells for 24 h and immunostaining for the presence viral NP. Images captured at 100 times magnification. The indicated significance (determined by one-way ANOVA) and percentage reduction in viral RNA detection are relative to the corresponding controls. (F,G) TG was superior to remdesivir (RDV) in blocking OC43 (F) and influenza A virus (G) replication. The A549 cells were primed with the indicated TG, 0.3 µM RDV or DMSO control for 30 min, washed twice with PBS and infected with 0.01 MOI of CoV OC43 or 1.0 MOI of USSR H1N1 virus for 2 h, after which cells were washed again with PBS and incubated in serum-free media for TG primed cells, or in media in the continuous presence of RDV. At 24, 48 and 72 hpi, viral RNA extraction was performed on the collected supernatants followed by one-step reverse transcription qPCR to detect the relative copy number of OC43 replicase polyprotein 1ab RNA or influenza M-gene RNA, based on the relative Ct method. The indicated significance is relative to the corresponding RDV-treated cells based on 2-way ANOVA Dunnett’s (F) or Tukey’s (G) multiple comparisons test. (H) RDV and TG treatments had no adverse effect on cell viability. A549 cells were treated continuously with RDV, or for 30 min with the indicated TG or DMSO, washed, cultured overnight and subjected to cell viability assay (CellTiter-Glo 2.0 Cell Viability Assay, Promega). The indicated significance was determined by one-way ANOVA relative to the DMSO control. (I) Selectivity index (CC50/EC50) of TG in OC43 inhibition was estimated at between 7072 and 9227. EC90 = 0.02622 μM. MRC5 cells were primed with TG (0 to 91 µM) for 30 min, washed twice with PBS and culture in DMEM Glutamax with 10% FCS and 1% P/S overnight. Cell viability assay (CC50) was performed with CellTiter-Glo 2.0 Cell Viability Assay (Promega). The effective or inhibition TG dose response (EC50) was based on priming of MRC5 cells with the indicated concentrations of TG (0 to 0.5 µM) for 30 min followed by PBS washing and infection with OC43 at 0.01 MOI. Three days post-infection, the supernatants were harvested for RNA extraction and one-step reverse-transcription qPCR to quantify the presence of viral RNA (polyprotein 1ab RNA). CC = cell cytotoxicity; EC = effective concentration. ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
![Viruses 13 00234 g004]()
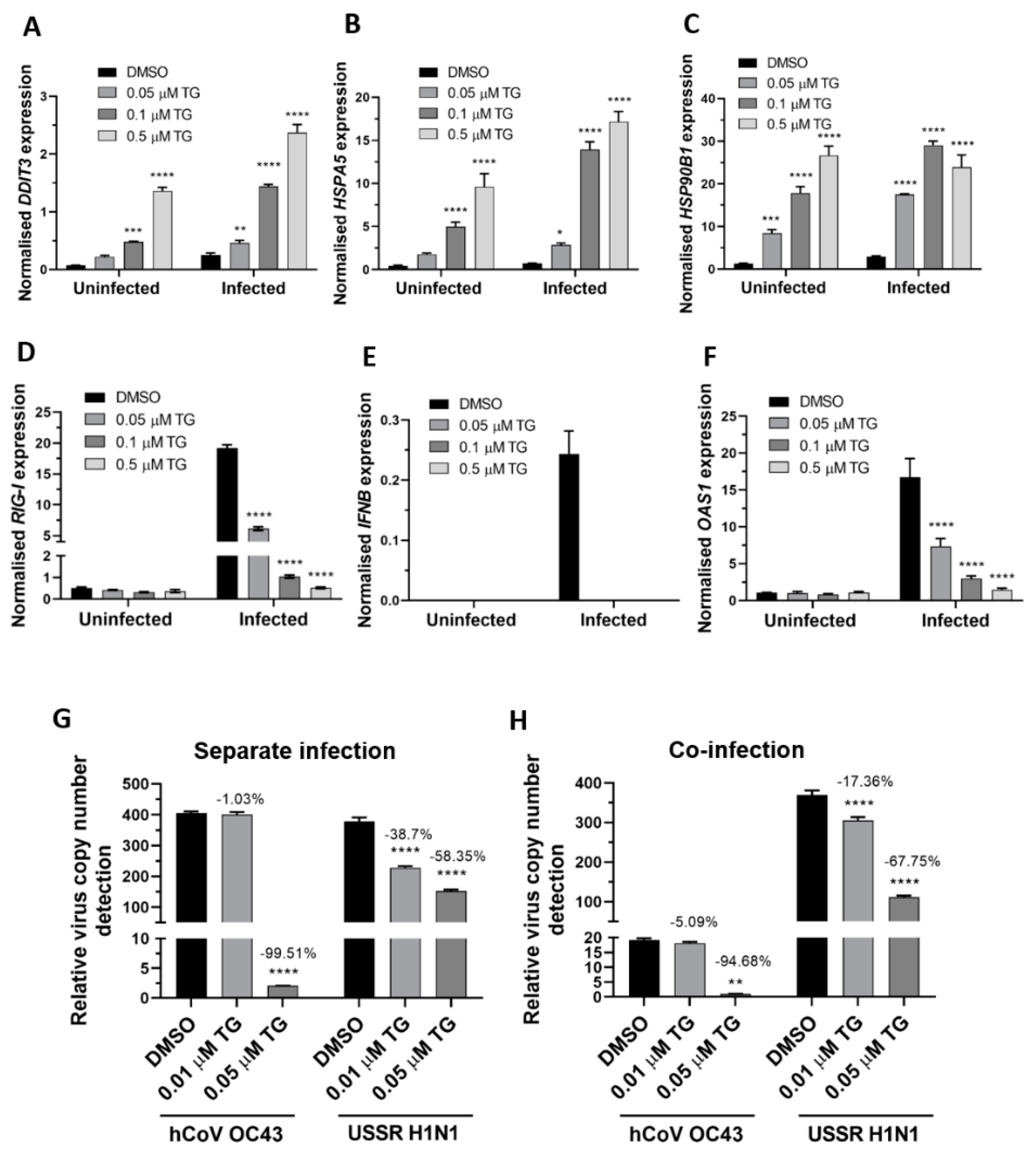
Figure 5.
TG priming of A549 cells increases expression of ER stress genes basally and during OC43 infection, attenuates induction of the RIG-I signalling-associated genes during infection, and inhibits co-infection with the OC43 and influenza viruses. (A–C). TG priming appeared to stimulate ER stress gene expression in a dose-dependent manner. (D–F) TG attenuated the induction of the RIG-I-associated genes during infection. A549 cells were primed with TG for 30 min, washed twice with PBS and infected with OC43 at 0.5 MOI for 3 h; thereafter cells were washed again with PBS and cultured for 24 h in serum-free media, after which cell lysates were harvested for RNA extraction and cDNA conversion for qPCR of (A) DDIT3, (B) HSPA5, (C) HSP90B1, (D) RIG-I, (E) IFNB and (F) OAS1. All expression was normalised to 18s rRNA. Significance is relative to the corresponding DMSO control based on 2-way ANOVA (Tukey’s multiple comparisons). (G,H) In A549 cells, TG inhibited the replication of OC43 virus and USSR H1N1 virus in separate virus infection or in co-infection. Cells were primed with TG for 30 min, washed twice with PBS and infected with the OC43 virus and USSR H1N1 virus at 0.01 and 1.5 MOI, respectively (based on FFAs), as single virus infection or co-infection for 3 h; after which cells were again washed twice with PBS and incubated in serum-free media. Culture media were harvested at 48 hpi for viral RNA extraction followed by one-step reverse transcription qPCR to detect the relative copy number of OC43 replicase polyprotein 1ab RNA and USSR H1N1 M-gene RNA. The indicated significance is based on 2-way ANOVA Tukey’s multiple comparisons and the percentage reduction in viral RNA detection are relative to the corresponding DMSO control. All assays were in triplicates and were performed three times. * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
Figure 5.
TG priming of A549 cells increases expression of ER stress genes basally and during OC43 infection, attenuates induction of the RIG-I signalling-associated genes during infection, and inhibits co-infection with the OC43 and influenza viruses. (A–C). TG priming appeared to stimulate ER stress gene expression in a dose-dependent manner. (D–F) TG attenuated the induction of the RIG-I-associated genes during infection. A549 cells were primed with TG for 30 min, washed twice with PBS and infected with OC43 at 0.5 MOI for 3 h; thereafter cells were washed again with PBS and cultured for 24 h in serum-free media, after which cell lysates were harvested for RNA extraction and cDNA conversion for qPCR of (A) DDIT3, (B) HSPA5, (C) HSP90B1, (D) RIG-I, (E) IFNB and (F) OAS1. All expression was normalised to 18s rRNA. Significance is relative to the corresponding DMSO control based on 2-way ANOVA (Tukey’s multiple comparisons). (G,H) In A549 cells, TG inhibited the replication of OC43 virus and USSR H1N1 virus in separate virus infection or in co-infection. Cells were primed with TG for 30 min, washed twice with PBS and infected with the OC43 virus and USSR H1N1 virus at 0.01 and 1.5 MOI, respectively (based on FFAs), as single virus infection or co-infection for 3 h; after which cells were again washed twice with PBS and incubated in serum-free media. Culture media were harvested at 48 hpi for viral RNA extraction followed by one-step reverse transcription qPCR to detect the relative copy number of OC43 replicase polyprotein 1ab RNA and USSR H1N1 M-gene RNA. The indicated significance is based on 2-way ANOVA Tukey’s multiple comparisons and the percentage reduction in viral RNA detection are relative to the corresponding DMSO control. All assays were in triplicates and were performed three times. * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
![Viruses 13 00234 g005]()
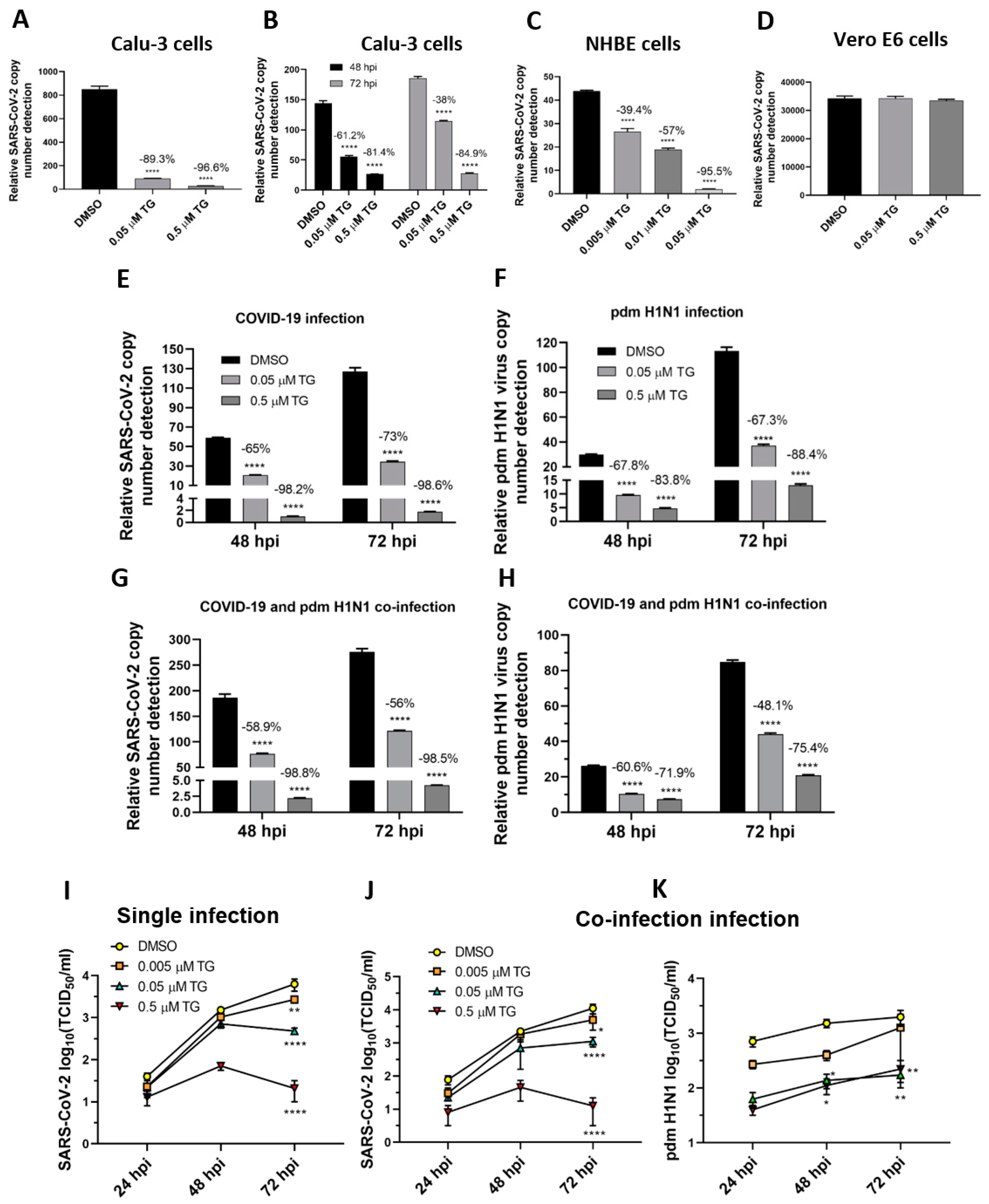
Figure 6.
TG effectively blocks progeny SARS-CoV-2 production in single-virus infection and in co-infection with the pdm H1N1 virus. (A,C,D) Pre-infection TG priming of Calu-3 and NHBE cells, but not Vero E6 cells, effectively inhibited progeny virus output. Cells were primed with TG as indicated for 30 min, washed twice with PBS and infected with SARS-CoV-2 at 0.01 MOI for 3 h; after which cells were again washed twice with PBS and incubated in serum-free media, supplemented with 0.2 µg/mL TPCK trypsin. Viral RNA extraction was performed on culture media at 72 hpi. (B) Priming with TG at 24 hpi blocked replication of SARS-CoV-2 in Calu-3 cells. Cells were first infected with SARS-CoV-2 at 0.01 MOI for 24 h, then primed with the indicated TG for 30 min, washed 3 times with PBS and incubated in serum-free media. Viral RNA extraction was performed on culture media at 48 and 72 hpi. All RNA isolated above were subjected to one-step reverse transcription qPCR to detect the relative copy number of SARS-CoV-2 replicase polyprotein 1ab RNA, based on the relative Ct method. Pre-infection priming of Calu-3 cells with TG inhibited separate infection of (E) SARS-CoV-2 and (F) the pdm H1N1 virus, and (G,H) co-infection with both viruses. Cells were primed with TG or DMSO for 30 min, infected with the corresponding virus at 0.01 MOI for 2 h, washed 3 times with PBS and incubated in serum-free media. At indicated time points post-infection, the media of infected cells were sampled for viral RNA extraction to perform one-step reverse transcription qPCR to detect the relative copy number of SARS-CoV-2 replicase polyprotein 1ab RNA (E,G) and influenza M-gene RNA (F,H). (I–K) Media samples taken at 24, 48 and 72 hpi from similarly infected cultures were used to detect viable progeny virus by TCID50 virus titration in Vero cells (displayed as mean ± SEM). (I) In single-virus infection with SARS-CoV-2, TG exhibited dose-dependent virus inhibition. In co-infection with SARS-CoV-2 and pdm H1N1 virus, TG was just as capable in inhibiting SARS-CoV-2 (J) and pdm H1N1 virus (K) at the same time. The indicated significance is based on 2-way ANOVA Tukey’s multiple comparisons test and percentage viral RNA change relative to the corresponding DMSO control. * p < 0.05, ** p < 0.01 and **** p < 0.0001.
Figure 6.
TG effectively blocks progeny SARS-CoV-2 production in single-virus infection and in co-infection with the pdm H1N1 virus. (A,C,D) Pre-infection TG priming of Calu-3 and NHBE cells, but not Vero E6 cells, effectively inhibited progeny virus output. Cells were primed with TG as indicated for 30 min, washed twice with PBS and infected with SARS-CoV-2 at 0.01 MOI for 3 h; after which cells were again washed twice with PBS and incubated in serum-free media, supplemented with 0.2 µg/mL TPCK trypsin. Viral RNA extraction was performed on culture media at 72 hpi. (B) Priming with TG at 24 hpi blocked replication of SARS-CoV-2 in Calu-3 cells. Cells were first infected with SARS-CoV-2 at 0.01 MOI for 24 h, then primed with the indicated TG for 30 min, washed 3 times with PBS and incubated in serum-free media. Viral RNA extraction was performed on culture media at 48 and 72 hpi. All RNA isolated above were subjected to one-step reverse transcription qPCR to detect the relative copy number of SARS-CoV-2 replicase polyprotein 1ab RNA, based on the relative Ct method. Pre-infection priming of Calu-3 cells with TG inhibited separate infection of (E) SARS-CoV-2 and (F) the pdm H1N1 virus, and (G,H) co-infection with both viruses. Cells were primed with TG or DMSO for 30 min, infected with the corresponding virus at 0.01 MOI for 2 h, washed 3 times with PBS and incubated in serum-free media. At indicated time points post-infection, the media of infected cells were sampled for viral RNA extraction to perform one-step reverse transcription qPCR to detect the relative copy number of SARS-CoV-2 replicase polyprotein 1ab RNA (E,G) and influenza M-gene RNA (F,H). (I–K) Media samples taken at 24, 48 and 72 hpi from similarly infected cultures were used to detect viable progeny virus by TCID50 virus titration in Vero cells (displayed as mean ± SEM). (I) In single-virus infection with SARS-CoV-2, TG exhibited dose-dependent virus inhibition. In co-infection with SARS-CoV-2 and pdm H1N1 virus, TG was just as capable in inhibiting SARS-CoV-2 (J) and pdm H1N1 virus (K) at the same time. The indicated significance is based on 2-way ANOVA Tukey’s multiple comparisons test and percentage viral RNA change relative to the corresponding DMSO control. * p < 0.05, ** p < 0.01 and **** p < 0.0001.
![Viruses 13 00234 g006]()
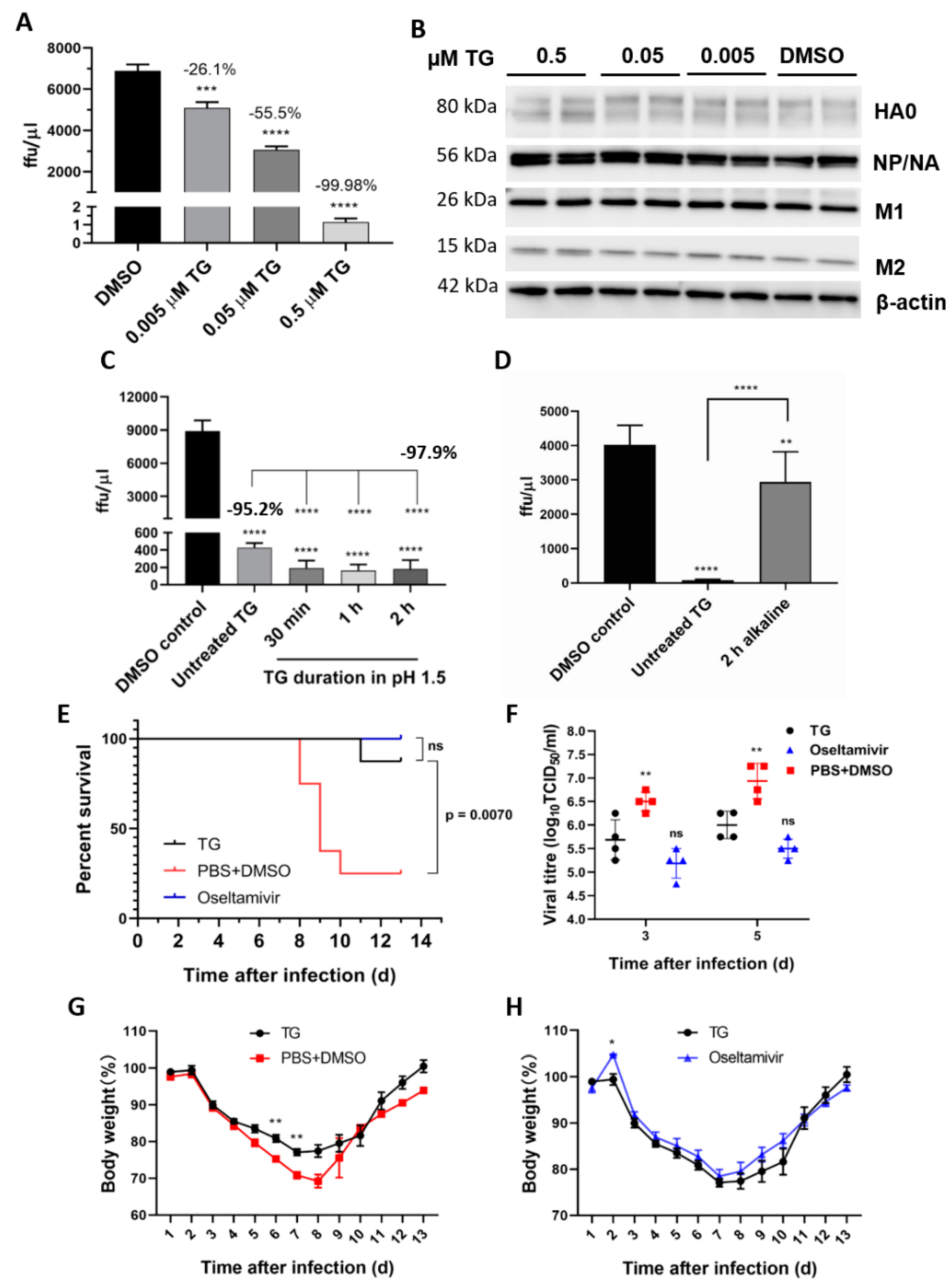
Figure 7.
TG post-translationally inhibits the influenza A virus, is acid-stable and therapeutically protects mice in a lethal virus challenge. (A) A sharp reduction in influenza progeny production from the TG-primed NPTr cells was (B) accompanied by no detectable reduction in viral proteins. Cells were primed with TG for 30 min, washed twice with PBS and infected with USSR virus at 0.5 MOI for 2 h; after which cells were again washed 3 times with PBS and incubated in serum-free media, supplemented with TPCK trypsin at 0.2 µgl/mL, for 24 h. (A) A focus forming assay was performed with the corresponding media samples to determine viable virus production (ffu/µL). The indicated significance is based on one-way ANOVA (Dunnett’s multiple comparisons) and the percentage viable progeny relative to the corresponding DMSO control. (B) Viral proteins, including those processed through the ER–Golgi apparatus (HA, NA and M2), showed no reduction in TG-primed cells. (C,D) Antiviral activity of TG was stable in acidic but not alkaline condition. (C) TG used was first incubated in pH 1.5 (in 30 mM hydrochloric acid) for different durations, as indicated, and neutralised with sodium hydroxide before being applied to cells for 30 min at a 0.5 μM final concentration. (D) TG used was first incubated in pH 12.0 (10 mM sodium hydroxide) for 2 h and neutralised with hydrochloric acid before being applied to cells for 30 min at a 0.5 μM final concentration. Twenty-four hours post-infection with the USSR H1N1 virus at 0.5 MOI, the infected culture media were used in 6 h focus forming assays to immuno-detect the viral NP to determine the progeny virus output (ffu/μL). Unless otherwise indicated, the significance is based on one-way ANOVA Tukey’s multiple comparison and percentage virus reduction are in relation to the corresponding DMSO control. (E–H) Therapeutic protection of TG in a lethal influenza virus challenge in mice. Each BALB/c mouse (n = 8 per group) in a group was intranasally infected with 3 MLD50 of PR8/H1N1 virus. Each mouse was then dosed orally once a day with TG (1.5 μg/kg/day), oseltamivir (45 mg/kg/day) or PBS+DMSO for 5 days; the first dose was given at 12 hpi. Survival rates (E), viral titres in the lung by TCID50 assays (F) and body weight changes (G,H) were recorded over 14 d. Each time point represents the mean ± SEM. The Kaplan–Meier method was used for survival analysis. Significance indicated relative to the corresponding TG group. * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
Figure 7.
TG post-translationally inhibits the influenza A virus, is acid-stable and therapeutically protects mice in a lethal virus challenge. (A) A sharp reduction in influenza progeny production from the TG-primed NPTr cells was (B) accompanied by no detectable reduction in viral proteins. Cells were primed with TG for 30 min, washed twice with PBS and infected with USSR virus at 0.5 MOI for 2 h; after which cells were again washed 3 times with PBS and incubated in serum-free media, supplemented with TPCK trypsin at 0.2 µgl/mL, for 24 h. (A) A focus forming assay was performed with the corresponding media samples to determine viable virus production (ffu/µL). The indicated significance is based on one-way ANOVA (Dunnett’s multiple comparisons) and the percentage viable progeny relative to the corresponding DMSO control. (B) Viral proteins, including those processed through the ER–Golgi apparatus (HA, NA and M2), showed no reduction in TG-primed cells. (C,D) Antiviral activity of TG was stable in acidic but not alkaline condition. (C) TG used was first incubated in pH 1.5 (in 30 mM hydrochloric acid) for different durations, as indicated, and neutralised with sodium hydroxide before being applied to cells for 30 min at a 0.5 μM final concentration. (D) TG used was first incubated in pH 12.0 (10 mM sodium hydroxide) for 2 h and neutralised with hydrochloric acid before being applied to cells for 30 min at a 0.5 μM final concentration. Twenty-four hours post-infection with the USSR H1N1 virus at 0.5 MOI, the infected culture media were used in 6 h focus forming assays to immuno-detect the viral NP to determine the progeny virus output (ffu/μL). Unless otherwise indicated, the significance is based on one-way ANOVA Tukey’s multiple comparison and percentage virus reduction are in relation to the corresponding DMSO control. (E–H) Therapeutic protection of TG in a lethal influenza virus challenge in mice. Each BALB/c mouse (n = 8 per group) in a group was intranasally infected with 3 MLD50 of PR8/H1N1 virus. Each mouse was then dosed orally once a day with TG (1.5 μg/kg/day), oseltamivir (45 mg/kg/day) or PBS+DMSO for 5 days; the first dose was given at 12 hpi. Survival rates (E), viral titres in the lung by TCID50 assays (F) and body weight changes (G,H) were recorded over 14 d. Each time point represents the mean ± SEM. The Kaplan–Meier method was used for survival analysis. Significance indicated relative to the corresponding TG group. * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
![Viruses 13 00234 g007]()
Table 1.
Primer sequences.
Table 1.
Primer sequences.
| Gene | Sense Primer (5′–3′) | Antisense Primer (5′–3′) |
|---|
| 18S ribosomal RNA (universal) | ACGGCTACCACATCCAAGGA | CCAATTACAGGGCCTCG-AAA |
| F gene (RSV) | CAAGAACTGACAGAGGATGGTACTG | CATGTTTCAGCTTGTGGGAAGA |
| L gene (RSV) | AACACTTATCCTTCTTTGTTGGAACTTA | GCAACCGAAACTCACGATAGAAA |
| M-gene (RSV) | ACTCAAGAAGTGCAGTGCTAGCA | AAGGACACATTAGCGCATATGGT |
| RIG-I (human) | GAAGGCATTGACATTGCACAGT | TGGTTTGGATCATTTTGATGACA |
| M-gene (USSR H1N1) | AGATGAGCCTTCTAACCGAGGTCG | TGCAAAAACATCTTCAA-GTCTCTG |
| M-gene (pdm H1N1) | AGATGAGTCTTCTAACCGAGGTCG | TGCAAAGACACTTTCCA-GTCTCTG |
| Orf1ab (SARS-CoV-2) | CCGATCATCAGCACATCTAGGTT | GACAAGGCTCTCCATCT-TACCTTT |
| Orf1ab (OC43) | GCCAGGGACGTGTTGTATCC | TTGATCTTCGACATTGTGACCTATG |


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}